调整原子顺序
背景
使用高斯建模得到的
代码
py
import numpy as np
from pywfn.base import Mole
from pywfn.reader import LogReader
from pywfn.writer import GjfWriter
path = r"c:\Users\11032\Desktop\gfile\碳硼烷\B12H12_21_wfn.log"
reader = LogReader(path)
mole = Mole(reader)
map_dick = {}
for i, sym in enumerate(mole.atoms.syms()):
if sym != "B":
continue
old_dist = 100
map_dick[i] = 0
for j, sym in enumerate(mole.atoms.syms()):
if sym != "H":
continue
dist = np.linalg.norm(mole.atoms.xyzs[i] - mole.atoms.xyzs[j])
if dist < old_dist:
map_dick[i] = j
old_dist = dist
syms = []
xyzs = []
for key, val in map_dick.items():
print(key, val)
syms.append(mole.atoms.syms[key])
xyzs.append(mole.atoms.xyzs[key])
syms.append(mole.atoms.syms[val])
xyzs.append(mole.atoms.xyzs[val])
writer = GjfWriter()
writer.syms = syms
writer.xyzs = xyzs
writer.job = "m062x 6-311g nosymm pop=full gfinput"
writer.charge = -2
writer.multip = 1
writer.save("test.gjf")结果
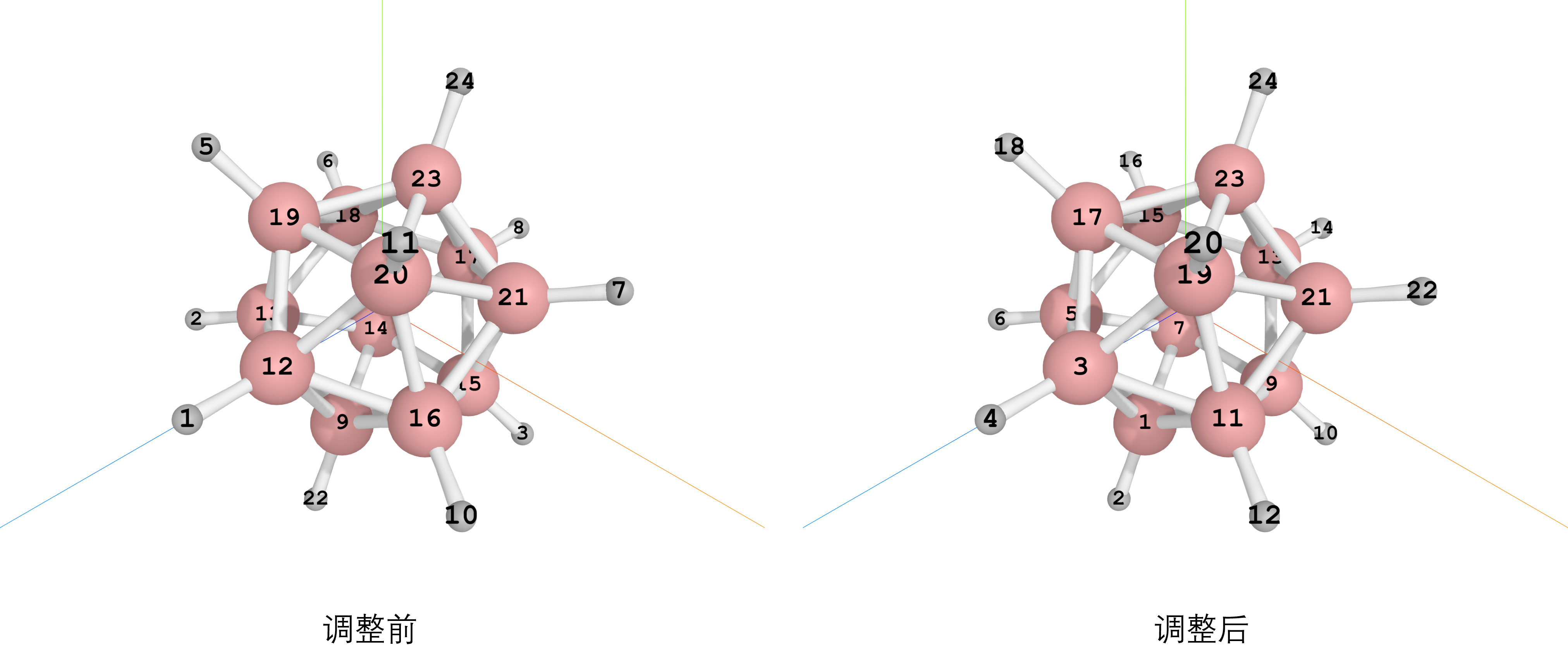 调整之后所有的B-H的原子索引是相邻的了
调整之后所有的B-H的原子索引是相邻的了